Assembling data¶
What constitutes a good assembly?
How to estimate assembly quality
Co-assembly
 These instructions assume you are using the EBI Training Virual Machines. If you’re running this on your own computer instead, remember that some file paths will be different. In particular,
These instructions assume you are using the EBI Training Virual Machines. If you’re running this on your own computer instead, remember that some file paths will be different. In particular, /home/training/ will be /home/<yourusername>.
Assembly and Co-assembly¶
Learning Objectives - in the following exercises you will
learn how to perform a metagenomic assembly and to start some basic
analysis of the output. Subsequently, we will demonstrate the
application of co-assembly. Note, due to the complexity of metagenomics
assembly, we will only be investigating very simple example datasets as
these often take days of CPU time and 100s of GB of memory. Thus, do not
think that there is an issue with the assemblies.
Once you have quality filtered your sequencing reads, you may want to perform de novo assembly in addition to, or as an alternative to a read-based analyses. The first step is to assemble your sequences into contigs. There are many tools available for this, such as MetaVelvet, metaSPAdes, IDBA-UD, MEGAHIT. We generally use metaSPAdes, as in most cases it yields the best contig size statistics (i.e. more continguous assembly) and has been shown to be able to capture high degrees of community diversity (Vollmers, et al. PLOS One 2017). However, you should consider the pros and cons of different assemblers, which not only includes the accuracy of the assembly, but also their computational overhead. Compare these factors to what you have available. For example, very diverse samples with a lot of sequence data uses a lot of memory with SPAdes. In the following practicals we will demonstrate the use of metaSPAdes on a small sample and the use of MEGAHIT for performing co-assembly.
Let’s first change to the working directory where we will be running the analyses:
cd /home/training/Data/Assembly/files
To run metaspades you would execute the following commands (but don’t run it now!):
mkdir assembly
metaspades.py -t 4 --only-assembler -m 10 -1 reads/oral_human_example_1_splitaa_kneaddata_paired_1.fastq -2 reads/oral_human_example_1_splitaa_kneaddata_paired_2.fastq -o assembly
 Since the assembly process would take ~1h we are just going to analyse the output present in assembly.bak. Let’s look at the contigs.fasta file.
Since the assembly process would take ~1h we are just going to analyse the output present in assembly.bak. Let’s look at the contigs.fasta file.
For this, take the first 40 lines of the sequence and perform a blast search at NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi, choose Nucleotide:Nucleotide from the set of options). Leave all other options as default on the search page. To select the first 40 lines of the assembly perform the following:
head -41 assembly.bak/contigs.fasta
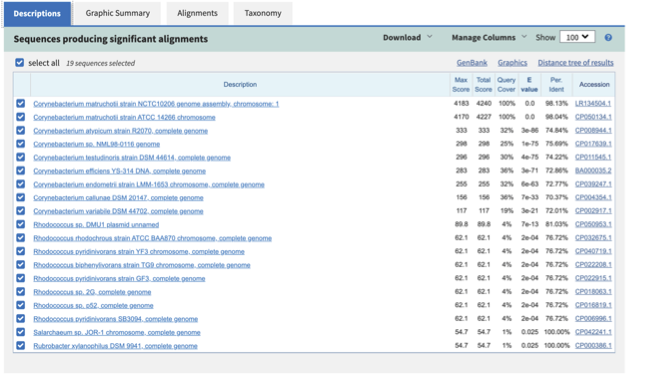
 Which species do you think this sequence may be coming from?
Does this make sense as a human oral bacteria? Are you surprised by this
result at all?
Which species do you think this sequence may be coming from?
Does this make sense as a human oral bacteria? Are you surprised by this
result at all?
Now let us consider some statistics about the entire assembly
assembly_stats assembly.bak/scaffolds.fasta
This will output two simple tables in JSON format, but it is
fairly simple to read. There is a section that corresponds to the
scaffolds in the assembly and a section that corresponds to the contigs.
What is the length of longest and shortest contigs?
What is the N50 of the assembly? Given that are input
sequences were ~150bp long paired-end sequences, what does this tell you
about the assembly?
N50 is a measure to describe the quality of assembled genomes
that are fragmented in contigs of different length. We can apply this
with some caution to metagenomes, where we can use it to crudely assess
the contig length that covers 50% of the total assembly. Essentially
the longer the better, but this only makes sense when thinking about
alike metagenomes. Note, N10 is the minimum contig length to cover 10
percent of the metagenome. N90 is the minimum contig length to cover 90
percent of the metagenome.
Bandage (a Bioinformatics Application for Navigating De novo
Assembly Graphs Easily), is a program that creates interactive
visualisations of assembly graphs. They can be useful for finding
sections of the graph, such as rRNA, or to try to find parts of a
genome. Note, you can install Bandage on your local system. With
Bandage, you can zoom and pan around the graph and search for sequences,
plus much more. The following guide allows you to look at the assembly
graph. Normally, I would recommend looking at the ‘
assembly_graph.fastg, but our assembly is quite fragmented, so we will
load up the assembly_graph_after_simplification.gfa.
At the terminal, type
Bandage
In the the Bandage GUI perform the following
Select File -> Load graph
Navigate to Home -> Data -> Assembly -> files -> assembly.bak and open the file assembly_graph_after_simplification.gfa
Once loaded, you need to draw the graph. To do so, under the “Graph drawing” panel on the left side perform the following:
Set Scope to ‘Entire graph’
The click on Draw graph
Use the sliders in the main panel to move around and look at
each distinct part of the assembly graph.
Can you find any large, complex parts of the graph? If so,
what do they look like.
In this particular sample, we believe that strains related to
the species Rothia dentocariosa, a Gram-positive, round- to rod-shaped
bacteria that is part of the normal community of microbes residing in
the mouth and respiratory tract, should be present in our sample. While
this is a tiny dataset, lets try to see if there is evidence for this
genome. To do so, we will search the R. dentocariosa genome against
the assembly graph.
To do so, go to the ‘BLAST’ panel on the left side of the GUI.
Step 1 - Select ‘Create/view BLAST search’, this will open a new window
Step 2 - Select ‘build Blast database’
Step 3 - Load from FASTA file. Navigate to the genome folder: Home -> Data -> Assembly -> files -> genome and select GCA_000164695.fasta
Step 4 - Modify the BLAST filters to 95% identity
Step 5 - Run BLAST
Step 6 - Close this window
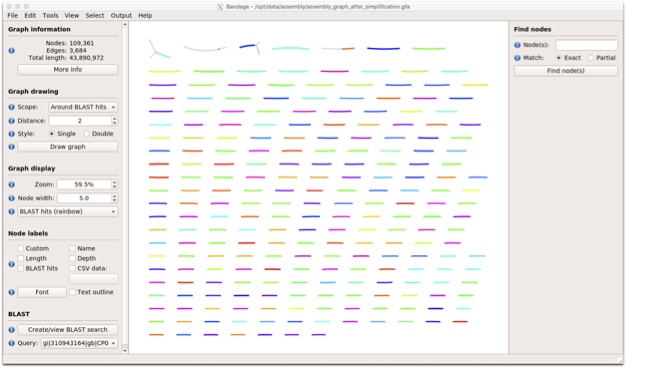
To visualise just these hits, go back to “Graph drawing” panel.
Set Scope to ‘Around BLAST hits’
Set Distance 2
The click on ‘Draw graph’
You should then see something like this:
In the following steps of this exercise, we will look at
performing co-assembly of multiple datasets. Each should take about 15-20 min. In case you do not manage to finish these on time, the directory coassembly.bak contains all the expected results.
First, we need to make sure the output directories we are going to create do not already exist (MEGAHIT cannot overwrite existing directories). Run:
rm -rf coassembly/assembly*
Then, perform the coassemblies with MEGAHIT, as follows:
megahit -1 reads/oral_human_example_1_splitac_kneaddata_paired_1.fastq -2 reads/oral_human_example_1_splitac_kneaddata_paired_2.fastq -o coassembly/assembly1 -t 4 --k-list 23,51,77
megahit -1 reads/oral_human_example_1_splitac_kneaddata_paired_1.fastq,reads/oral_human_example_1_splitab_kneaddata_paired_1.fastq -2 reads/oral_human_example_1_splitac_kneaddata_paired_2.fastq,reads/oral_human_example_1_splitab_kneaddata_paired_2.fastq -o coassembly/assembly2 -t 4 --k-list 23,51,77
megahit -1 reads/oral_human_example_1_splitab_kneaddata_paired_1.fastq,reads/oral_human_example_1_splitac_kneaddata_paired_1.fastq,reads/oral_human_example_1_splitaa_kneaddata_paired_1.fastq -2 reads/oral_human_example_1_splitab_kneaddata_paired_2.fastq,reads/oral_human_example_1_splitac_kneaddata_paired_2.fastq,reads/oral_human_example_1_splitaa_kneaddata_paired_2.fastq -o coassembly/assembly3 -t 4 --k-list 23,51,77
You should now have three different assemblies, let us compare the results.
assembly_stats coassembly/assembly1/final.contigs.fa
assembly_stats coassembly/assembly2/final.contigs.fa
assembly_stats coassembly/assembly3/final.contigs.fa
How do these assemblies differ to the one generated previously with metaSPAdes? Which one do you think is best?